•A bisulfite-free method detects 5fC at the single-cell and single-base resolution •5fC is highly dynamic and enriched in functionally important genomic regions •Intrinsic 5fC heterogeneity is revealed for mouse early embryos, mEpiSC, and mESCs •Emergence of promoter 5fC production precedes the upregulation of gene expression
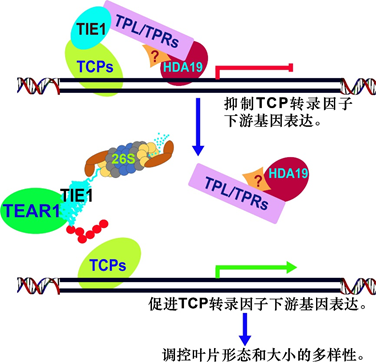
The developmental plasticity of leaf size and shape is important for leaf function and plant survival. However, the mechanisms by which plants form diverse leaves in response to environmental conditions are not well understood. Here, we identified TIE1-ASSOCIATED RING-TYPE E3 LIGASE1 (TEAR1) and found that it regulates leaf development by promoting the degradation of TCP INTERACTOR-CONTAINING EAR MOTIF PROTEIN1 (TIE1), an important repressor of CINCINNATA (CIN)-like TEOSINTE BRANCHED1/CYCLOIDEA/PCF (TCP) transcription factors, which are key for leaf development. TEAR1 contains a typical C3H2C3-type RING domain and has E3 ligase activity. We show that TEAR1 interacts with the TCP repressor TIE1, which is ubiquitinated in vivo and degraded by the 26S proteasome system. We demonstrate that TEAR1 is colocalized with TIE1 in nuclei and negatively regulates TIE1 protein levels. Overexpression of TEAR1 rescued leaf defects caused by TIE1 overexpression, whereas disruption of TEAR1 resulted in leaf phenotypes resembling those caused by TIE1 overexpression or TCP dysfunction. Deficiency in TEAR partially rescued the leaf defects of TCP4 overexpression line and enhanced the wavy leaf phenotypes of jaw-5D. We propose that TEAR1 positively regulates CIN-like TCP activity to promote leaf development by mediating the degradation of the TCP repressor TIE1.
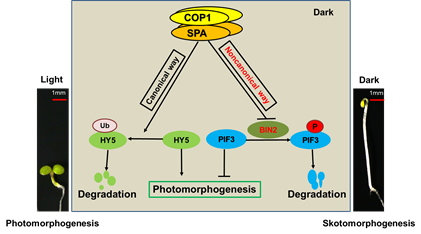
Initially discovered as a central repressor of the seedling photomorphogenesis in plants, and later characterized in animals, the E3 ligase CONSTITUTIVELY PHOTOMORPHOGENIC 1 (COP1) has been shown to have conserved biochemical activities in eukaryotes. In this study, we show that to coordinately promote skotomorphogenesis, the COP1/ SUPPRESSOR of phyA-105 (SPA) complex represses the activity of the GSK3-like kinase BRASSINOSTEROID-INSENSITIVE 2 (BIN2) in a nonproteolytic manner to stabilize the key repressor of photomorphogenesis PHYTOCHROME INTERACTING FACTOR 3 (PIF3). The COP1/SPA-BIN2-PIF3 module represents an additional layer of COP1/SPA regulation in Arabidopsis and is essential for shaping the plants when grown in the dark.
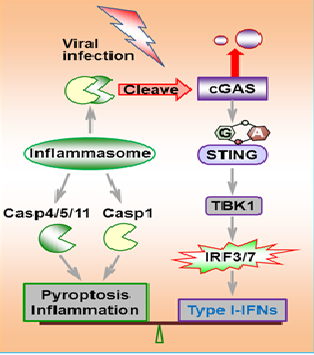
Viral infection triggers host innate immune responses that result in the production of various cytokines including type I interferons (IFN), activation of inflammasomes, and programmed cell death of the infected cells. Tight control of inflammatory cytokine production is crucial for the triggering of an effective immune response that can resolve the infection without causing host pathology. In examining the inflammatory response of Asc−/− and Casp1−/− macrophages, we found that deficiency in these molecules resulted in increased IFN production upon DNA virus infection, but not RNA virus challenge. Investigation of the underlying mechanism revealed that upon canonical and non-canonical inflammasome activation, caspase-1 interacted with cyclic GMP-AMP (cGAMP) synthase (cGAS), cleaving it and dampening cGAS-STING-mediated IFN production. Deficiency in inflammasome signaling enhanced host resistance to DNA virus in vitro and in vivo, and this regulatory role extended to other inflammatory caspases. Thus, inflammasome activation dampens cGAS-dependent signaling, suggesting cross-regulation between intracellular DNA-sensing pathways.
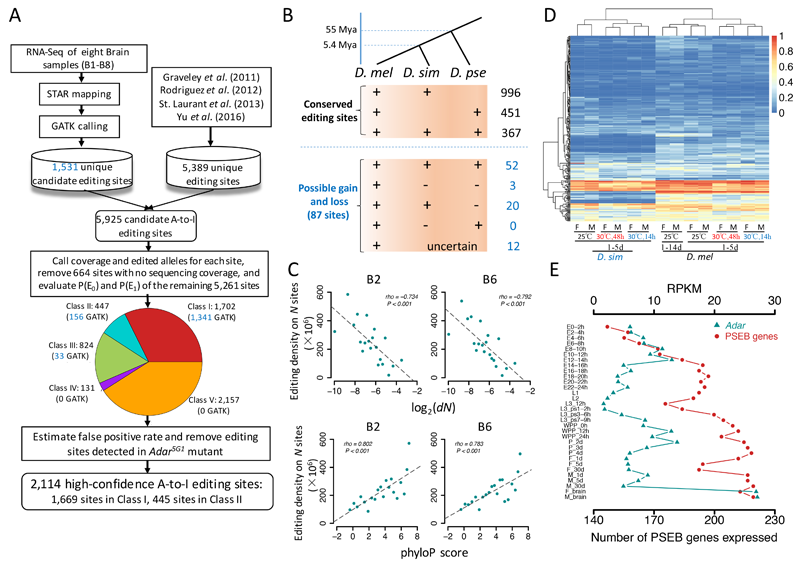
Adenosine-to-inosine (A-to-I) RNA editing is an evolutionarily conserved mechanism that alters RNA sequences at the co-transcriptional or post-transcriptional level. RNA editing is hypothesized to facilitate adaptation in that it expands the transcriptomic and proteomic diversity. However, evidence for adaptation of RNA editing at the whole editome level is still lacking. In this study we systematically identified A-to-I RNA editing sites in female and male brains of three Drosophila species at different temperatures. With evolutionary analysis from different perspectives, we provide lines of evidence to demonstrate that the nonsynonymous editing sites in Drosophila brains are generally adaptive. The signals of adaptation for the editing sites are significantly enriched in genes related to chemical and electrical neurotransmission. We show that the RNA editing events might interplay with gene expression plasticity in temperature stress responses. Furthermore, we demonstrated that the expression level of Adar, together with the expression profiles of a set of genes that have editing sites favored by natural selection, were important in shaping the overall selective patterns of the global editomes at different developmental stages (or tissues) of D. melanogaster. Altogether our results support the hypothesis that A-to-I editing provides a driving force for adaptive evolution in Drosophila from different aspects.
The tiger is most recognized for its dark stripes against an orange background. Less well known are three other pelage color variants: white, golden and stripeless snow white. The white tiger is a polymorphism that was first seen among wild Bengal tigers (P. t. tigris) in India, with white fur and sepia brown stripes. The golden tiger, also first sighted in the jungle in India, has a blonde color tone with pale golden fur and red-brown rather than black stripes. The snow white tiger is almost completely white, with faint to nearly nonexistent narrow stripes on the trunk and diluted sepia brown rings on the tail. During the past century, the number of wild tigers in the world has severely declined primarily due to anthropogenic reasons, and except for the wild-type orange tigers, all other variants now exist only in captivity.
Many insects, including mosquitoes, planthoppers, aphids and leafhoppers, are the hosts of bacterial symbionts and the vectors for transmitting viral pathogens. In general, symbiotic bacteria can indirectly affect viral transmission by enhancing immunity and resistance to viruses in insects. Whether symbiotic bacteria can directly interact with the virus and mediate its transmission has been unknown. Here, we show that an insect symbiotic bacterium directly harbours a viral pathogen and mediates its transovarial transmission to offspring. We observe rice dwarf virus (a plant reovirus) binding to the envelopes of the bacterium Sulcia, a common obligate symbiont of leafhoppers, allowing the virus to exploit the ancient oocyte entry path of Sulcia in rice leafhopper vectors. Such virus–bacterium binding is mediated by the specific interaction of the viral capsid protein and the Sulcia outer membrane protein. Treatment with antibiotics or antibodies against Sulcia outer membrane protein interferes with this interaction and strongly prevents viral transmission to insect offspring. This newly discovered virus–bacterium interaction represents the first evidence that a viral pathogen can directly exploit a symbiotic bacterium for its transmission. We believe that such a model of virus–bacterium communication is a common phenomenon in nature.
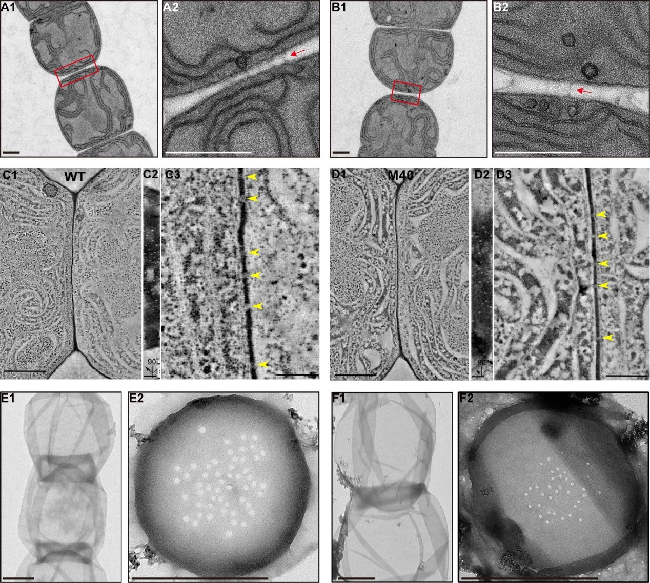
The filamentous cyanobacterium Anabaena has become a widely studied model to determine the molecular mechanisms involved in establishing and maintaining the pattern of heterocyst differentiation in response to the removal of fixed nitrogen from the environment. Heterocysts develop from vegetative cells, usually spaced about 10 cells apart, converting an oxic cell capable of division into an anoxic factory for nitrogen fixation that does not divide. Genetic analysis to elucidate the mechanisms of intercellular material exchange between heterocysts and vegetative cells is in an early phase. Here we show that an amidase is involved in the function of channels that penetrate the rigid peptidoglycan walls that separate cells in the filaments.
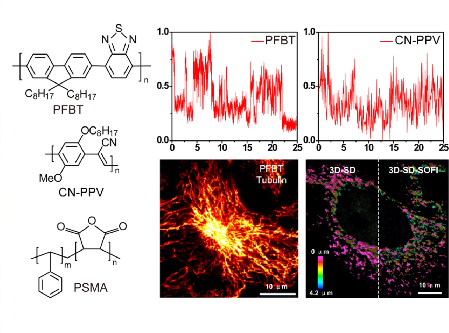
Two types of small photoblinking Pdots with high brightness, strong photostability, and favorable biocompatibility, are designed. Super-resolution optical fluctuation imaging is achieved using these Pdots. Imaging of subcellular structures demonstrates that these small photoblinking Pdots are outstanding probes for fast, long-term super-resolution fluorescence imaging.
Novel design and technical solutions for long-term two-photon imaging in awake monkey Stable expression of genetically encoded indicators in cortical neurons across months Hardware and software to stabilize two-photon images Simultaneous electrophysiology studies under two-photon imaging in awake monkeys
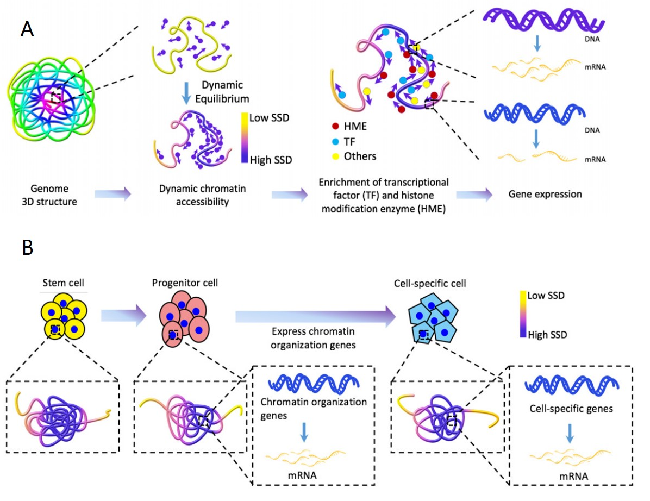
Chromatin three-dimensional (3D) structure plays critical roles in gene expression regulation by influencing locus interactions and accessibility of chromatin regions. Here we propose a Markov process model to derive a chromosomal equilibrium distribution of randomly-moving molecules as a functional consequence of spatially organized genome 3D structures. The model calculates steady-state distributions (SSD) from Hi-C data as quantitative measures of each chromatin region`s dynamic accessibility for transcription factors and histone modification enzymes. Different from other Hi-C derived features such as compartment A/B and interaction hubs, or traditional methods measuring chromatin accessibility such as DNase-seq and FAIRE-seq, SSD considers both chromatin–chromatin and protein–chromatin interactions. Through our model, we find that SSD could capture the chromosomal equilibrium distributions of activation histone modifications and transcription factors. Compared with compartment A/B, SSD has higher correlations with the binding of these histone modifications and transcription factors. In addition, we find that genes located in high SSD regions tend to be expressed at higher level. Furthermore, we track the change of genome organization during stem cell differentiation, and propose a two-stage model to explain the dynamic change of SSD and gene expression during differentiation, where chromatin organization genes first gain chromatin accessibility and are expressed before lineage-specific genes do. We conclude that SSD is a novel and better measure of dynamic chromatin activity and accessibility.
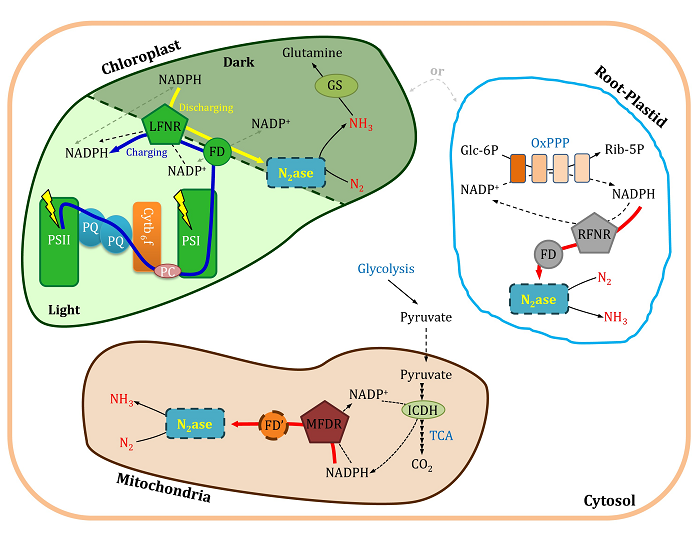
Engineering nitrogenase into cereal crops requires detailed understanding of the components required for efficient nitrogen fixation. We have used a synthetic biology modular approach to evaluate components from chloroplast, root plastids, and mitochondria that function as electron donors to both conventional Mo nitrogenase and the alternative Fe nitrogenase systems. The knowledge obtained in this study not only identifies electron-transfer components from plant organelles that can be used to support nitrogenase activity, but also is likely to enable reduction of the number of target genes required to engineer nitrogen fixation in plants.