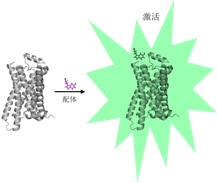
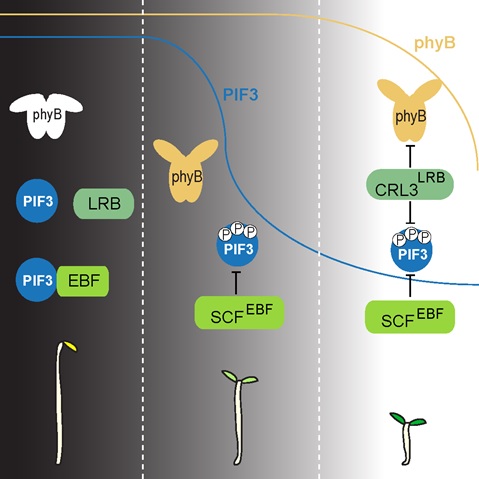
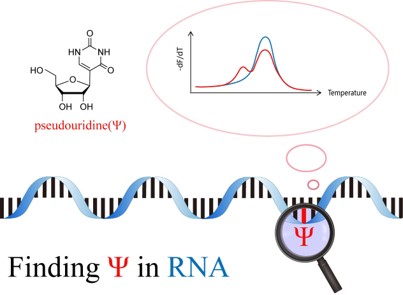
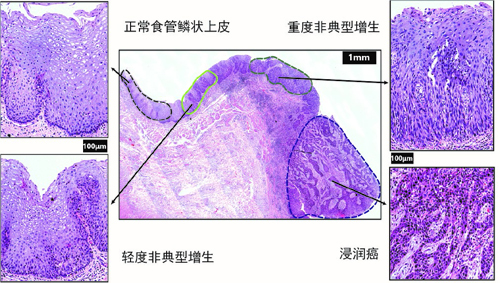
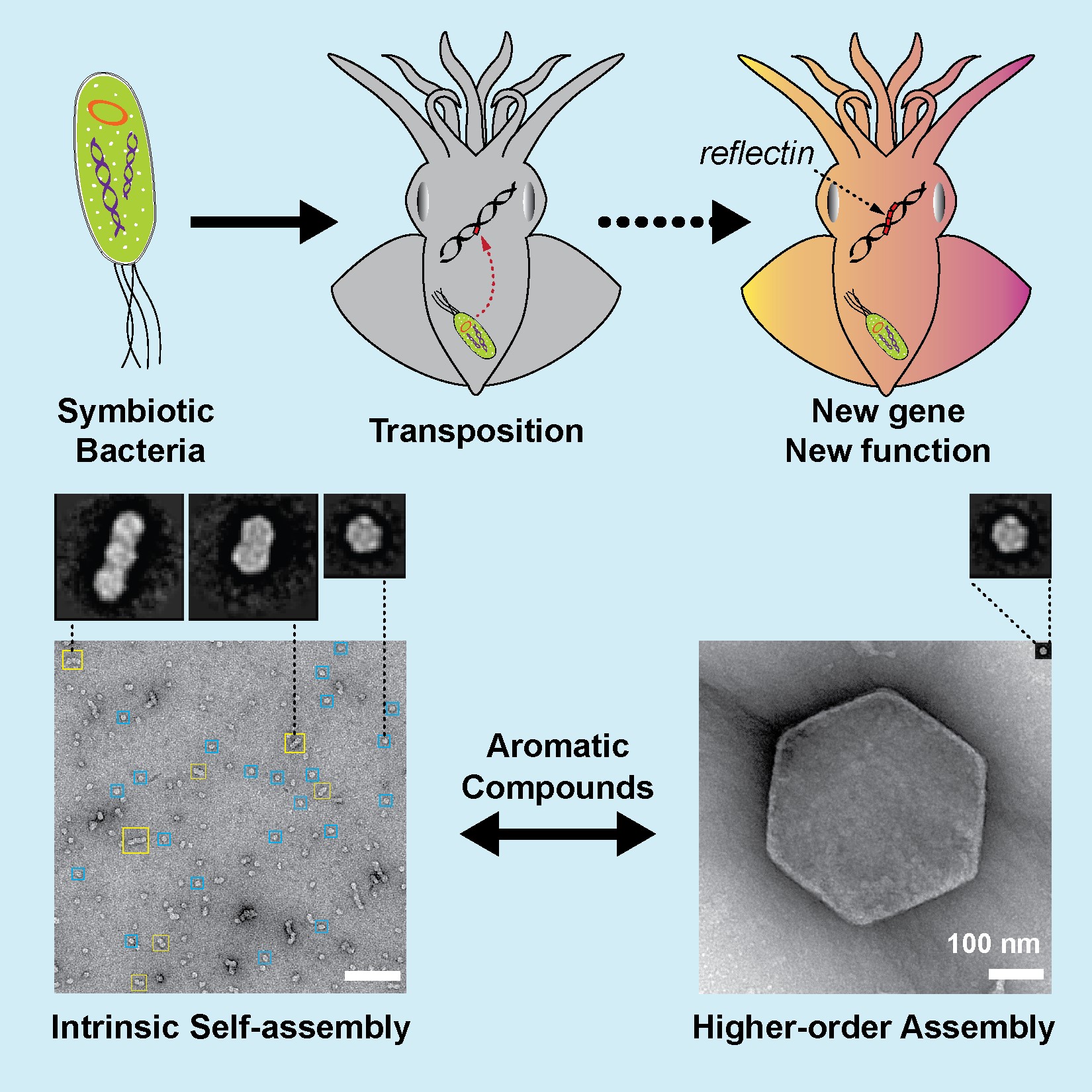
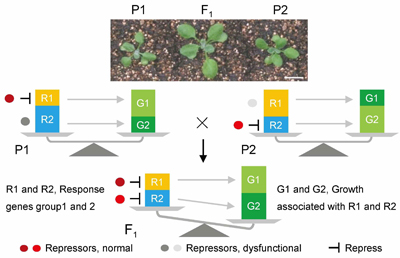
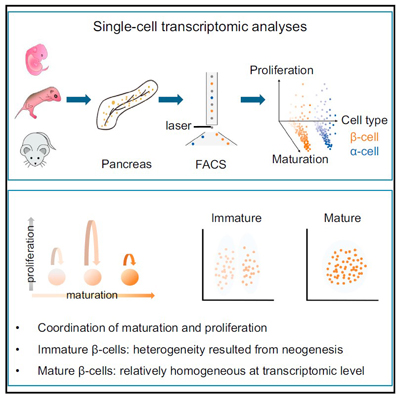
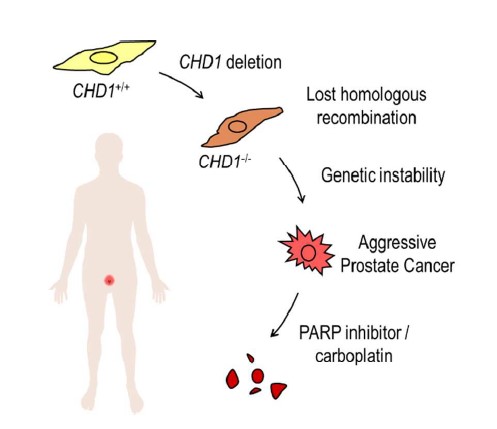
 Jul 10,2017
Jul 10,2017
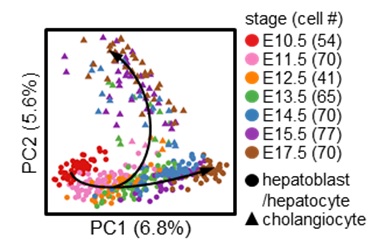
How the bi-potential hepatoblasts differentiate into hepatocytes and cholangiocytes remains unclear. Here, using single-cell transcriptomic analysis of hepatoblasts, hepatocytes, and cholangiocytes sorted from E10.5 to E17.5 mouse embryos, we found that hepatoblast-to-hepatocyte differentiation occurred gradually followed a linear default pathway. As more cells became fully differentiated hepatocytes, the number of proliferating cells decreased. Surprisingly, the proliferating and quiescent hepatoblasts exhibited homogeneous differentiation states at a given developmental stage. This unique feature enabled us to combine the single-cell and bulk-cell analyses to define the precise timing of the hepatoblast-to-hepatocyte transition, which occurs between E13.5 and E15.5. In contrast to hepatocyte development at almost all levels, hepatoblast-to-cholangiocyte differentiation underwent a sharp detour from the default pathway. New cholangiocyte generation occurred continuously between E11.5 and E14.5, but their maturation states at a given developmental stage were heterogeneous. Even more surprising, the number of proliferating cells increased as more progenitor cells differentiated into mature cholangiocytes. Based on an observation from the single-cell analysis, we also discovered that the protein kinase C (PKC)/mitogen-activated protein kinase (MAPK) signaling pathway promoted cholangiocyte maturation. Conclusions: Our studies have defined distinct pathways for hepatocyte and cholangiocyte development in vivo, which are critically important for understanding basic liver biology and developing effective strategies to induce stem cells to differentiate towards specific hepatic cell fates in vitro. This article is protected by copyright. All rights reserved.
 Jun 16,2017
Jun 16,2017
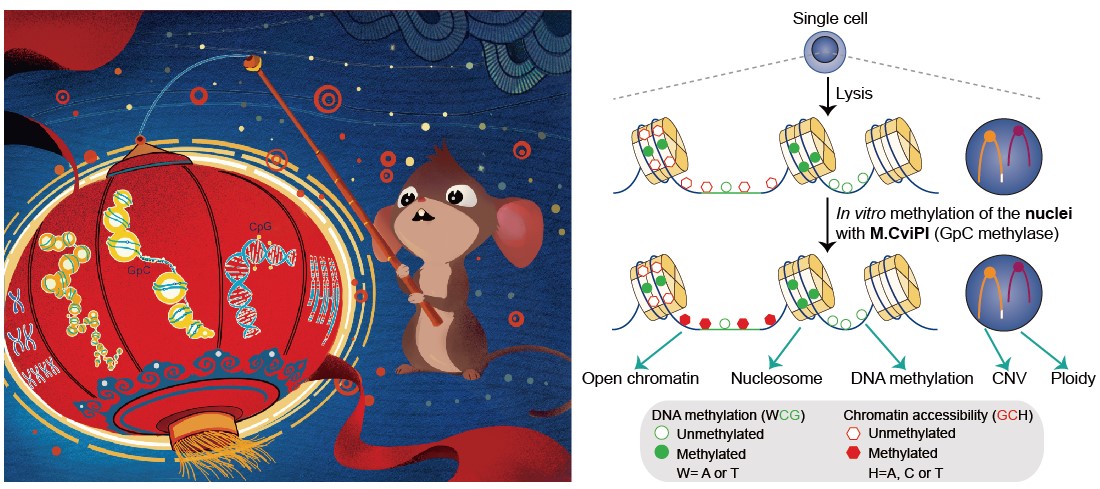
Single-cell epigenome sequencing techniques have recently been developed. However, the combination of different layers of epigenome sequencing in an individual cell has not yet been achieved. Here, we developed a single-cell multi-omics sequencing technology (single-cell COOL-seq) that can analyze the chromatin state/nucleosome positioning, DNA methylation, copy number variation and ploidy simultaneously from the same individual mammalian cell. We used this method to analyze the reprogramming of the chromatin state and DNA methylation in mouse preimplantation embryos. We found that within < 12 h of fertilization, each individual cell undergoes global genome demethylation together with the rapid and global reprogramming of both maternal and paternal genomes to a highly opened chromatin state. This was followed by decreased openness after the late zygote stage. Furthermore, from the late zygote to the 4-cell stage, the residual DNA methylation is preferentially preserved on intergenic regions of the paternal alleles and intragenic regions of maternal alleles in each individual blastomere. However, chromatin accessibility is similar between paternal and maternal alleles in each individual cell from the late zygote to the blastocyst stage. The binding motifs of several pluripotency regulators are enriched at distal nucleosome depleted regions from as early as the 2-cell stage. This indicates that the cis-regulatory elements of such target genes have been primed to an open state from the 2-cell stage onward, long before pluripotency is eventually established in the ICM of the blastocyst. Genes may be classified into homogeneously open, homogeneously closed and divergent states based on the chromatin accessibility of their promoter regions among individual cells. This can be traced to step-wise transitions during preimplantation development. Our study offers the first single-cell and parental allele-specific analysis of the genome-scale chromatin state and DNA methylation dynamics at single-base resolution in early mouse embryos and provides new insights into the heterogeneous yet highly ordered features of epigenomic reprogramming during this process.