 Mar 01,2018
Mar 01,2018
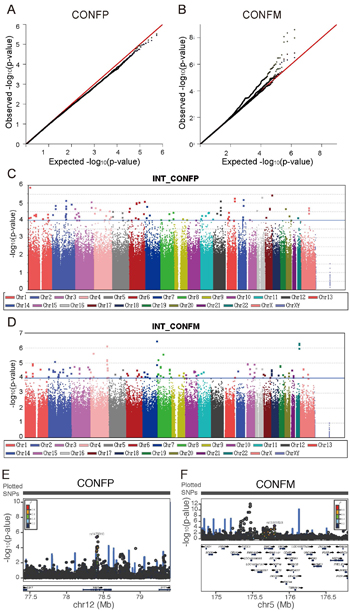
Social conformity is fundamental to human societies and has been studied for more than six decades, but our understanding of its mechanisms remains limited. Individual differences in conformity have been attributed to social and cultural environmental influences, but not to genes. Here we demonstrate a genetic contribution to conformity after analyzing 1,140 twins and single-nucleotide polymorphism (SNP)-based studies of 2,130 young adults. A two-step genome-wide association study (GWAS) revealed replicable associations in 9 genomic loci, and a meta-analysis of three GWAS with a sample size of ~2,600 further confirmed one locus, corresponding to the NAV3 (Neuron Navigator 3) gene which encodes a protein important for axon outgrowth and guidance. Further multi-level (haplotype, gene, pathway) GWAS strongly associated genes including NAV3, PTPRD (protein tyrosine phosphatase receptor type D), ARL10 (ADP ribosylation factor-like GTPase 10), and CTNND2 (catenin delta 2), with conformity. Magnetic resonance imaging of 64 subjects shows correlation of activation or structural features of brain regions with the SNPs of these genes, supporting their functional significance. Our results suggest potential moderate genetic influence on conformity, implicate several specific genetic elements in conformity and will facilitate further research on cellular and molecular mechanisms underlying human conformity.
 Dec 24,2017
Dec 24,2017
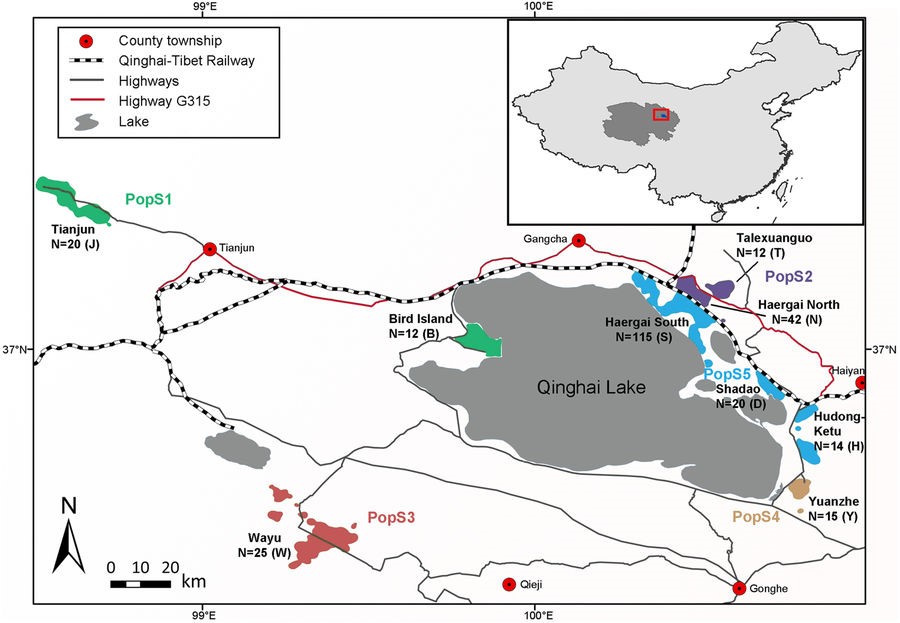
The Przewalski’s gazelle (Procapra przewalskii) is one of the most endangered ungulates in the world, with fewer than 2,000 individuals surviving in nine habitat fragments on the Qinghai-Tibet Plateau and isolated by human settlements and infrastructure. In particular, the Qinghai-Tibet railway, which crosses the largest part of the gazelle’s distribution, remains a major concern because of its potential to intensify landscape genetic differentiation. Here, using mtDNA sequencing and microsatellite genotyping to analyze 275 Przewalski’s gazelle samples collected throughout the range, we observed low level of genetic diversity (mtDNA π = 0.0033) and strong phylogeographic structure. Overall, the nine patches of gazelles can be further clustered into five populations, with a strong division between the eastern vs. western side of Qinghai Lake. Our study provides the first evidence of the genetic divergence between the Haergai North and Haergai South gazelle populations, corresponding to the recent construction of a wired enclosure along the Qinghai-Tibet railway less than ten years ago, an equivalent of five generations. Well-designed wildlife corridors across the railway along with long-term monitoring of the anthropogenic effects are therefore recommended to alleviate further habitat fragmentation and loss of genetic diversity in Przewalski’s gazelle.
 Dec 05,2017
Dec 05,2017
The Hi-C method is widely used to study the functional roles of the three-dimensional (3D) architecture of genomes. Here, we integrate Hi-C, whole-genome sequencing (WGS) and RNA-seq to study the 3D genome architecture of multiple myeloma (MM) and how it associates with genomic variation and gene expression. Our results show that Hi-C interaction matrices are biased by copy number variations (CNVs) and can be used to detect CNVs. Also, combining Hi-C and WGS data can improve the detection of translocations. We find that CNV breakpoints significantly overlap with topologically associating domain (TAD) boundaries. Compared to normal B cells, the numbers of TADs increases by 25% in MM, the average size of TADs is smaller, and about 20% of genomic regions switch their chromatin A/B compartment types. In summary, we report a 3D genome interaction map of aneuploid MM cells and reveal the relationship among CNVs, translocations, 3D genome reorganization, and gene expression regulation.