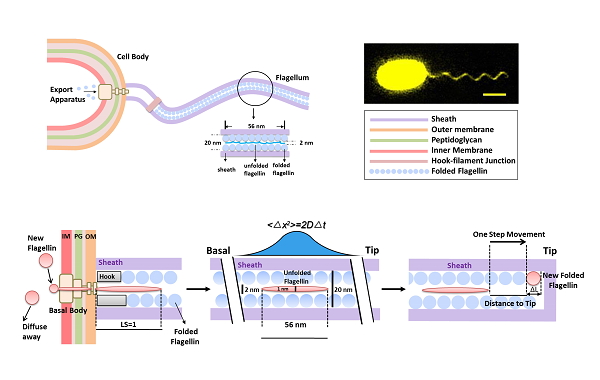
Bacterial flagella are extracellular filaments that drive swimming in bacteria. During motor assembly, flagellins are transported unfolded through the central channel in the flagellum to the growing tip. Here, we applied in vivo fluorescent imaging to monitor in real time the Vibrio alginolyticus polar flagella growth. The flagellar growth rate is found to be highly length-dependent. Initially, the flagellum grows at a constant rate (50 nm/min) when shorter than 1500 nm. The growth rate decays sharply when the flagellum grows longer, which decreases to ~9 nm/min at 7500 nm. We modeled flagellin transport inside the channel as a one-dimensional diffusive process with an injection force at its base. When the flagellum is short, its growth rate is determined by the loading speed at the base. Only when the flagellum grows longer does diffusion of flagellin become the rate-limiting step, dramatically reducing the growth rate. Our results shed new light on the dynamic building process of this complex extracellular structure.
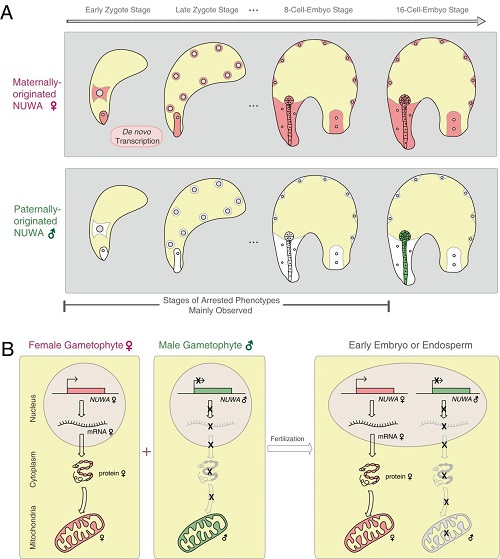
Imprinted genes are only found in mammals and flowering plants, and they express with parent-of-origin-specific patterns. Unlike in mammals, only a few imprinted genes was identified and functionally characterized in plants, and almost all of them encode nuclear proteins. Here, we identified NUWA as a new type of imprinted gene in Arabidopsis. NUWA has maternal-specific expression and controls the early seed development in Arabidopsis, with its encoded protein localizing to and functioning in the maternally inherited mitochondria. The discovery of NUWA reveals a unique mechanism of maternal control of mitochondrial function and adds an extra layer of complexity to nucleus-organelle coordination. It further indicates that, similar to mammals, imprinted genes in plants are also involved in various subcellular biological processes.
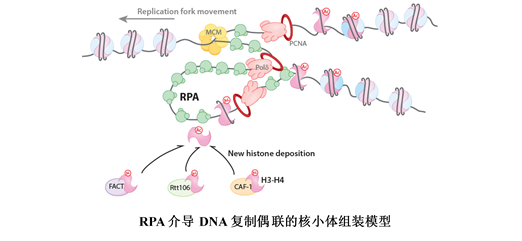
Nucleosomes removed from DNA to facilitate its replication must be replaced quickly to protect the genome. The epigenetic information stored on the parental nucleosomes must also be preserved on the daughter DNA strands. The replication protein A complex (RPA) is a critical component of the DNA replication machinery. RPA binds single-stranded DNA. Liu et al. show that RPA bound to a single-stranded DNA replication fork mimic recruits and promotes the assembly of H3-H4 histone tetramers onto adjacent double-stranded DNA. It also recruits specific H3-H4 chaperones, which facilitate the assembly of new nucleosomes.
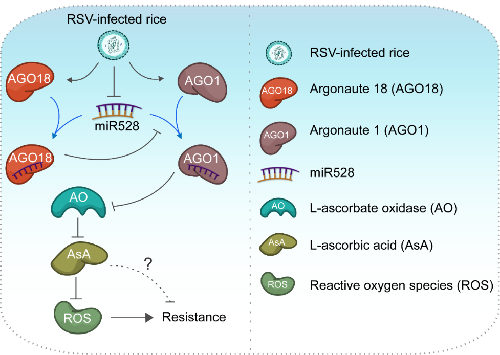
MicroRNAs (miRNAs) are key regulators of plant–pathogen interactions. Modulating miRNA function has emerged as a new strategy to produce virus resistance traits. However, the miRNAs involved in antiviral defence and the underlying mechanisms remain largely elusive. We previously demonstrated that sequestration by Argonaute (AGO) proteins plays an important role in regulating miRNA function in antiviral defence pathways. Here we reveal that cleavage-defective AGO18 complexes sequester microRNA528 (miR528) upon viral infection. We show that miR528 negatively regulates viral resistance in rice by cleaving L-ascorbate oxidase (AO) messenger RNA, thereby reducing AO-mediated accumulation of reactive oxygen species. Upon viral infection, miR528 becomes preferentially associated with AGO18, leading to elevated AO activity, higher basal reactive oxygen species accumulation and enhanced antiviral defence. Our findings reveal a mechanism in which antiviral defence is boosted through suppression of an miRNA that negatively regulates viral resistance. This mechanism could be manipulated to engineer virus-resistant crop plants.
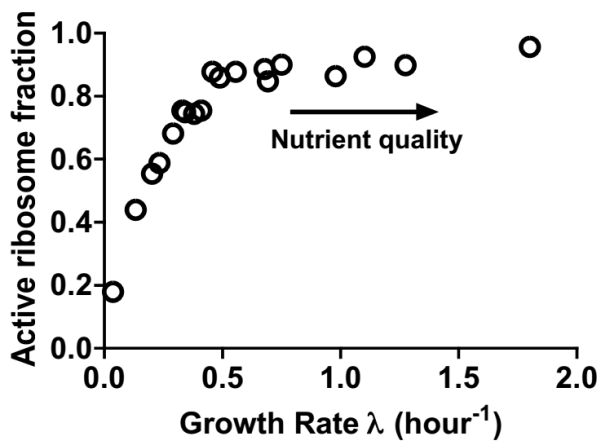
Bacterial growth significantly depends on protein synthesis catalyzed by ribosome. Ribosome translation elongation speed is a key factor determining the bacterial protein synthesis rate. However, existing methods for determining translation elongation speed have limited applications. Here we developed a simple and convenient method for measuring bacterial translation elongation speed based on LacZα complementation system. It enables the measurement of in vivo translation elongation speed of different individual genes. Tests related to ribosome translation elongation speed under various growth perturbations including different nutrient conditions, low temperature, a low-speed ribosome mutant, and fusidic acid treatment, were performed to quantitatively validate this method. Using this approach, we further found that nutrient starvation caused a remarkable slow-down of ribosome translation of Escherichia coli (E. coli). We also studied the dynamic change of translation elongation speed during the process of nutrient up-shift. This method will boost the quantitative understanding of bacterial ribosome translation capacity and growth.
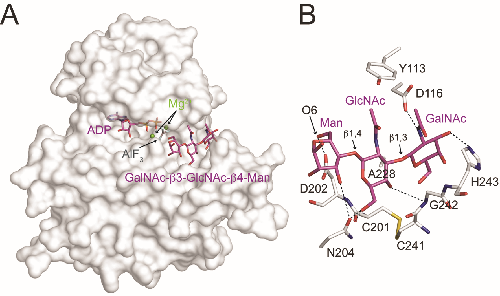
The ‘pseudokinase’ SgK196 is a protein O-mannose kinase (POMK) that catalyzes an essential phosphorylation step during biosynthesis of the laminin-binding glycan on α-dystroglycan. However, the catalytic mechanism underlying this activity remains elusive. Here we present the crystal structure of Danio rerio POMK in complex with Mg2+ ions, ADP, aluminum fluoride, and the GalNAc-β3-GlcNAc-β4-Man trisaccharide substrate, thereby providing a snapshot of the catalytic transition state of this unusual kinase. The active site of POMK is established by residues located in non-canonical positions and is stabilized by a disulfide bridge. GalNAc-β3-GlcNAc-β4-Man is recognized by a surface groove, and the GalNAc-β3-GlcNAc moiety mediates the majority of interactions with POMK. Expression of various POMK mutants in POMK knockout cells further validated the functional requirements of critical residues. Our results provide important insights into the ability of POMK to function specifically as a glycan kinase, and highlight the structural diversity of the human kinome.
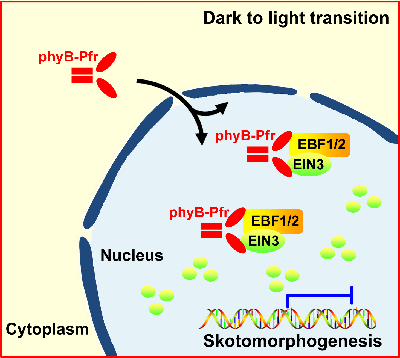
Plants germinating under subterranean darkness assume skotomorphogenesis, a developmental program strengthened by ethylene in response to mechanical pressure of soil. Upon reaching the surface, light triggers a dramatic developmental transition termed de-etiolation that requires immediate termination of ethylene responses. Here, we report that light activation of photoreceptor phyB results in rapid degradation of EIN3, the master transcription factor in the ethylene signaling pathway. As a result, light rapidly and efficiently represses ethylene actions. Specifically, phyB directly interacts with EIN3 in a light-dependent manner and also physically associates with F box protein EBFs. The light-activated association of phyB, EIN3, and EBF1/EBF2 proteins stimulates robust EIN3 degradation by SCFEBF1/EBF2 E3 ligases. We reveal that phyB manipulates substrate-E3 ligase interactions in a light-dependent manner, thus directly controlling the stability of EIN3. Our findings illustrate a mechanistic model of how plants transduce light information to immediately turn off ethylene signaling for de-etiolation initiation.
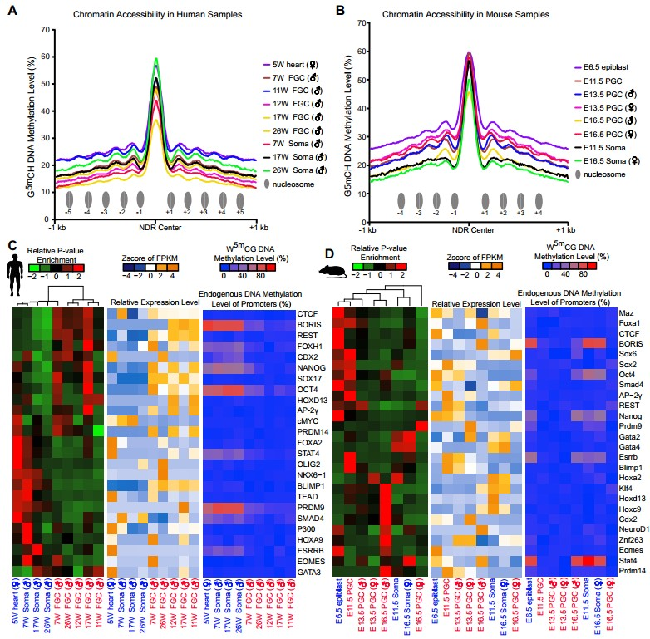
Chromatin remodeling is important for the epigenetic reprogramming of human primordial germ cells. However, the comprehensive chromatin state has not yet been analyzed for human fetal germ cells (FGCs). Here we use nucleosome occupancy and methylation sequencing method to analyze both the genome-wide chromatin accessibility and DNA methylome at a series of crucial time points during fetal germ cell development in both human and mouse. We find 116 887 and 137 557 nucleosome-depleted regions (NDRs) in human and mouse FGCs, covering a large set of germline-specific and highly dynamic regulatory genomic elements, such as enhancers. Moreover, we find that the distal NDRs are enriched specifically for binding motifs of the pluripotency and germ cell master regulators such as NANOG, SOX17, AP2γ and OCT4 in human FGCs, indicating the existence of a delicate regulatory balance between pluripotency-related genes and germ cell-specific genes in human FGCs, and the functional significance of these genes for germ cell development in vivo. Our work offers a comprehensive and high-resolution roadmap for dissecting chromatin state transition dynamics during the epigenomic reprogramming of human and mouse FGCs.
The recent development of reversibly switchable fluorescent proteins (RSFPs) has promoted reversible saturable optical fluorescence transitions (RESOLFT) nanoscopy as a general scheme for live-cell super-resolution imaging. However, continuous, long-term live-cell RESOLFT nanoscopy is still hindered mainly because of the unsatisfactory properties of existing RSFPs. In this work, we report GMars-Q, a monomeric RSFP with low residual off-state fluorescence and strong fatigue resistance attributed to a biphasic photobleaching process. We further demonstrate that GMars-Q is particularly suitable for long-term parallelized RESOLFT nanoscopy as it supports an order of magnitude longer imaging durations than existing RSFPs. The excellent photophysical properties of GMars-Q also suggest that it would be of general interest for other RESOLFT nanoscopic methods.
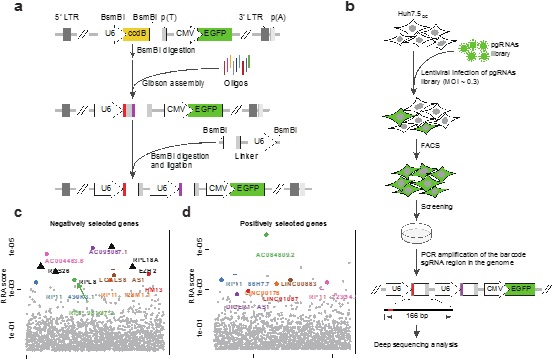
CRISPR–Cas9 screens have been widely adopted to analyze coding-gene functions, but high-throughput screening of non-coding elements using this method is more challenging because indels caused by a single cut in non-coding regions are unlikely to produce a functional knockout. A high-throughput method to produce deletions of non-coding DNA is needed. We report a high-throughput genomic deletion strategy to screen for functional long non-coding RNAs (lncRNAs) that is based on a lentiviral paired-guide RNA (pgRNA) library. Applying our screening method, we identified 51 lncRNAs that can positively or negatively regulate human cancer cell growth. We validated 9 of 51 lncRNA hits using CRISPR–Cas9-mediated genomic deletion, functional rescue, CRISPR activation or inhibition and gene-expression profiling. Our high-throughput pgRNA genome deletion method will enable rapid identification of functional mammalian non-coding elements.
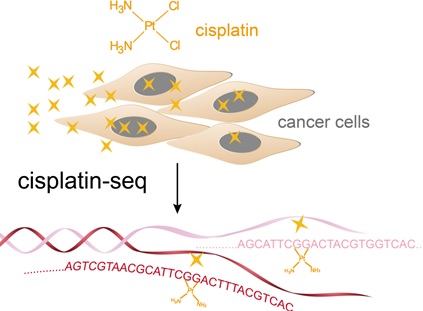
Cisplatin, one of the most widely used anticancer drugs, crosslinks DNA and ultimately induces cell death. However, the genomic pattern of cisplatin–DNA adducts has remained unknown owing to the lack of a reliable and sensitive genome-wide method. Herein we present “cisplatin-seq” to identify genome-wide cisplatin crosslinking sites at base resolution. Cisplatin-seq reveals that mitochondrial DNA is a preferred target of cisplatin. For nuclear genomes, cisplatin–DNA adducts are enriched within promoters and regions harboring transcription termination sites. While the density of GG dinucleotides determines the initial crosslinking of cisplatin, binding of proteins to the genome largely contributes to the accumulative pattern of cisplatin–DNA adducts.
 Dec 14,2016
Dec 14,2016